Scripting a DNA sequence viewer
By Bob Mesibov, published 08/11/2016 in Tutorials
SANITY WARNING: Please don't read this article unless you're a compulsive shell scripter, like me. Seriously. There are some excellent FOSS sequence viewers in the distro repositories, with great GUIs and lots of features. Why would you want to build a less capable viewer that only works in a terminal?
Still with me? OK, the script explained here is called 'DNA' and relies on up-to-date GNU utilities (e.g. GNU AWK) and BASH. As a command it takes 2 arguments: a (mandatory) filename and an (optional) starting nucleotide position. It works on fasta files with (ideally) no more than 52 aligned DNA sequences of equal length, like 'demo.fst', shown below. I've deliberately made 'demo.fst' ugly by putting single sequences on more than one line, inserting a blank line and using both lowercase and uppercase letters for the nucleotides. Not a problem, the script will handle it.
$ cat demo.fst
>H329_114_K1
TATCGGTACTTTATTTTTAATT--TGGTGCTTGAGCTG
GAATTATTGGATCTGCTTTGAGAGGGATAATTCGTATAGA
>H329_114_K2
TATCGGTACTTTATATTTAATTTTTGGTGCTTGAGCTG
GAATTATTGGATCTGCTTTGAGAGGAATAATTCGTATAGA
>H329_114_K3
TATCGGTACTTTATATTTGATTTTTGGTGCTTGAGCTG
GAATTATTGGATCTGCTTTGAGAGGGATAATTCGT-----
>H329_114_K4
TATCGCTACTTTATATTTGATTTTTGGTGCTTGAGCTG
GAATTATTGGATCTGCTTTGAGAGGGATAATTCGTATAGA
>H329_114_K5
TATCGGTACTTTATATTTGATTTTTGGTGCTTGAGCTG
GAATTATTGGATCTGCTTTGAGAGGGATAATTCGTATAGA
>H329_115_K1
----gctactttatatttgatttttggtgcttgagctg
gaattattggatctgctttgagagggataattcgtataga
>H329_115_K2
tatcggtactttatatttaatttttggtgcttgagctg
gaattattggatctgctttaagaggaataattcgtata--
>H329_115_K3
tatcggtactttatatttgatttttggtgcttgagctg
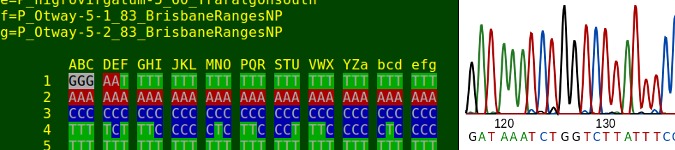
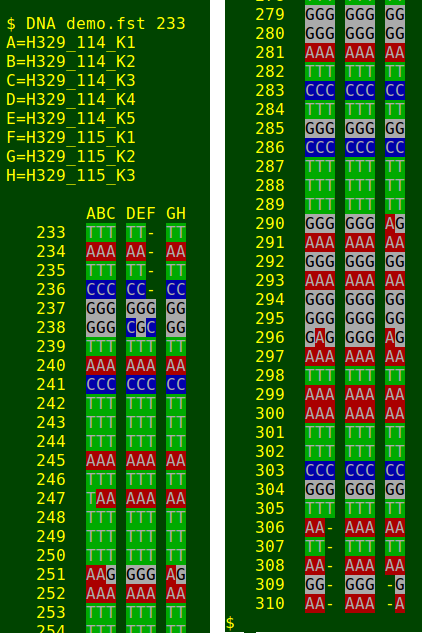
gaattattggatctgctttgagagggataattcgtataga'DNA' processes the file 'demo.fst' and displays the result in a terminal. In this case I've chosen the starting nucleotide position '233' (top and bottom of terminal output placed side by side for clarity):
The full 'DNA' script is shown at the bottom of the page. Because it's a wee bit complicated, I'll explain it in parts. The core of the script looks like this:
#!/bin/bash
A='\e[0;37;41mA'
C='\e[0;37;44mC'
G='\e[0;30;47mG'
T='\e[0;37;42mT'
f='\e[0m'
tr -d '\r' < "$1" > working
awk 'BEGIN {RS=">";FS="\n";OFS=""} NR>1 {print ">"$1; $1=""; print}' working > canon
sed -n '2~2p' canon | awk '{print toupper($0) > "sq"sprintf("%02d",NR)}'
for i in sq*; do fold -w1 "$i" > new"$i"; done
foo=$(paste -d "" newsq* | sed 's/.\{3\}/& /g' | nl \
| sed "s/A/\\$A\\$f/g;s/C/\\$C\\$f/g;s/G/\\$G\\$f/g;s/T/\\$T\\$f/g")
echo -e "$foo"
rm canon sq* newsq* working
exitFiddling with the FASTA
The first thing the script does is remove any Windows carriage returns from the fasta file with tr and save the result as the temp file 'working'. CR characters will bugger up almost any sort of text processing on the GNU command line. Exterminate!
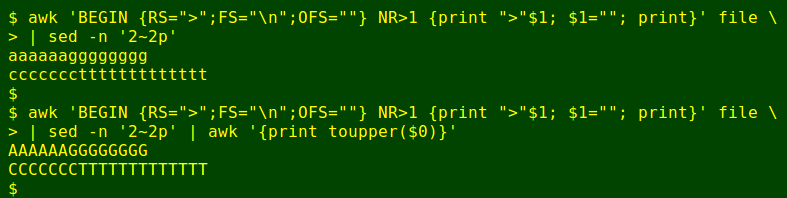
The display-building process in the script is designed to work with fasta files containing (for each sequence) a single ID line, like >H329_114_K1, followed by a single sequence line. In the 'demo.fst' file used above, these conditions aren't met. To get files into shape I use a very neat AWK command devised by UK coding wizard Bruce Horrocks. Here it's shown working on a simplified file:

The BEGIN statement sets the record separator RS to the '>' character. This means 'file' contains 3 records: [blank]>[xxx+sequence]>[yyy+sequence]. Each of the 3 records is initially divided into fields (FS) with a newline, so that, for example, field 1 of record 3 is 'yyy', field 2 is 'ccccccc' and field3 is 'ttttttttttttt'.
Next after BEGIN comes a condition: the command that follows only applies to records after record number 1 (NR>1), so the blank first record (before the first '>') is ignored. The first instruction in the command is to print the character '>' followed immediately by the first field, so '>xxx' gets printed.
The second instruction tells AWK that field 1 is redefined to be blank. Redefinition of a field causes AWK to redefine the whole record according to any earlier instructions. One of those earlier instructions (in BEGIN) is that the output field separator OFS is an empty string, and the sequence lines get concatenated by this simple redefinition of the record. This process is repeated for each record, and the rebuilt fasta file is saved (in the script) as a temp file, 'canon'.
Processing the sequences
The rebuilt fasta file 'canon' consists of alternating lines: ID, sequence, ID, sequence etc. I use sed to print just the sequence lines, which are then sent to AWK. AWK first makes sure that all the nucleotide codes are upper-case ('toupper'). To show how this works I'll again use the simplified file:
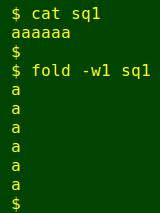
In the script, AWK then prints each sequence line separately to a new temp file called 'sq' followed by the line number. The line numbers are padded with a leading zero to 2 places with 'sprintf', as in 'sq01', 'sq09', 'sq10'; this padding is needed to preserve sequence order when the paste command is used later in the script. Next, each of the 'sq[line number]' temp files is processed with the fold -w1 command in a for loop to turn each horizontal run of nucleotides into a vertical run, as in this simplified example:
Each vertical run is saved as a temp file, 'newsq[line number]'.
Prettying the display
The next steps are more straightforward. The 'newsq' vertical runs are pasted together side by side with the paste separator option set to a blank (-d ""). A sed command is used to create a space between every 3 columns, for ease in viewing. The pasted-together-and-spaced runs are then numbered with nl. The resulting set of lines is sent to sed, which replaces each of the nucleotide letters with a coloured and highlighted version using ANSI escape codes. These coded escapes have been defined at the start of the script as variables, and note that the sed command uses double quotes so that these variables can be understood by the shell.
The prettied-up columns are saved as the variable 'foo', which is then echoed to the terminal with the '-e' option so that the escape backslashes can be interpreted. Finally, the temp files are deleted. That's the core script.
Specifying nucleotide position
A useful addition to the script is to have a second argument for the command that specifies a starting number for the first nucleotide position. I've made this second argument optional using an if loop (see script below). If no second argument is given ([[ -z "$2" ]]), the script defaults to numbering the positions from 1. I specify a starting position with the '-v' option for nl. Here's the modified script:
#!/bin/bash
A='\e[0;37;41mA'
C='\e[0;37;44mC'
G='\e[0;30;47mG'
T='\e[0;37;42mT'
f='\e[0m'
tr -d '\r' < "$1" > working
awk 'BEGIN {RS=">";FS="\n";OFS=""} NR>1 {print ">"$1; $1=""; print}' working > canon
sed -n '2~2p' canon | awk '{print toupper($0) > "sq"sprintf("%02d",NR)}'
for i in sq*; do fold -w1 "$i" > new"$i"; done
if [[ -z "$2" ]]; then
foo=$(paste -d "" newsq* | sed 's/.\{3\}/& /g' | nl \
| sed "s/A/\\$A\\$f/g;s/C/\\$C\\$f/g;s/G/\\$G\\$f/g;s/T/\\$T\\$f/g")
else
foo=$(paste -d "" newsq* | sed 's/.\{3\}/& /g' | nl -v "$2" \
| sed "s/A/\\$A\\$f/g;s/C/\\$C\\$f/g;s/G/\\$G\\$f/g;s/T/\\$T\\$f/g")
fi
echo -e "$foo"
rm canon sq* newsq* working
exitSequence labelling
A second tweak is to label the sequences. This is easy to do when the sequences are displayed horizontally, as in most sequence viewers, but it's a bit trickier when the sequences are vertical.
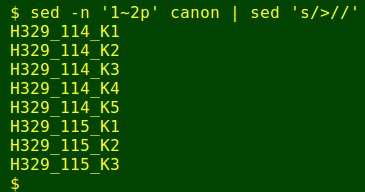
The first step is to build a legend for the sequences. I use sed to extract the ID lines from 'canon' and also to remove the leading '>' character:
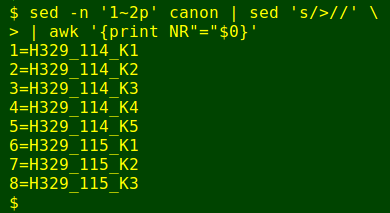
I then use AWK to build a legend with the sequences listed in the order they appear in the fasta file:
Meanwhile, I've created a BASH array called 'alphabet' containing the brace expansion of '{{A..Z},{a..z}}', which is the 26 uppercase letters in serial order followed by the 26 lowercase letters in serial order:

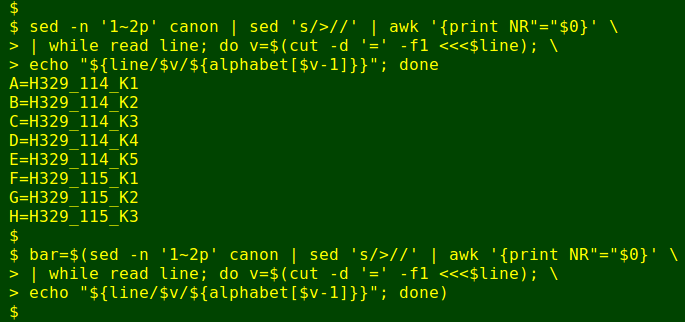
Now for some more BASH voodoo (see final script, below). I use the array 'alphabet' to replace (with BASH substitution) the numbers in the legend with their serial letter counterparts. I do this by working through the legend line by line with a while loop. The first character or characters in the line (the serial number ahead of the equals sign) is cut out and stored as the variable 'v'. BASH substitution is then done on the line, with 'v' replaced by a letter equivalent in the array. However, since BASH arrays number their elements from 0, the replacement characters are based on 1 less than the value of 'v'. For example, alphabet[0] is A. To replace '1' with 'A', I need to look up the value of alphabet[1-1]. It works:
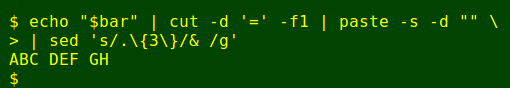
The revised legend is saved as the variable 'bar' and will be echoed straight to the terminal (see script below). To label the vertical sequence runs according to that legend I first extract the serial letters from 'bar' with cut and turn them into a horizontal row with paste, with sed again used to create a space every 3 letters:
How to align those letters over the first set of vertical columns? The trick is to remember that by default, nl right-justifies its numbers at the end of a block of 6 spaces, and follows the numbers with a tab. I can reproduce that behaviour exactly with a sed substitution:
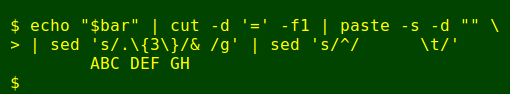
Because there are just 52 available labels (A>Z + a>z), the 'DNA' script can only label 52 sequences at a time. If there are more than 52 sequences in the fasta file, the extras will be displayed without labels (and will crowd the terminal!).
Disclaimer and full script
Like I said at the beginning, there are FOSS sequence viewers that put this shell script to shame, and I recommend their use if you're doing serious work with sequences. If you do use this script and your fasta file has very long sequences, it's best to pipe the result to less.
#!/bin/bash
A='\e[0;37;41mA'
C='\e[0;37;44mC'
G='\e[0;30;47mG'
T='\e[0;37;42mT'
f='\e[0m'
alphabet=($(echo {{A..Z},{a..z}}))
tr -d '\r' < "$1" > working
awk 'BEGIN {RS=">";FS="\n";OFS=""} NR>1 {print ">"$1; $1=""; print}' working > canon
bar=$(sed -n '1~2p' canon | sed 's/>//' | awk '{print NR"="$0}' \
| while read line; do v=$(cut -d '=' -f1 <<<$line); \
echo "${line/$v/${alphabet[$v-1]}}"; done)
echo "$bar"
echo ""
echo "$bar" | cut -d '=' -f1 | paste -s -d "" | sed 's/.\{3\}/& /g' | sed 's/^/ \t/'
sed -n '2~2p' canon | awk '{print toupper($0) > "sq"sprintf("%02d",NR)}'
for i in sq*; do fold -w1 "$i" > new"$i"; done
if [[ -z "$2" ]]; then
foo=$(paste -d "" newsq* | sed 's/.\{3\}/& /g' | nl \
| sed "s/A/\\$A\\$f/g;s/C/\\$C\\$f/g;s/G/\\$G\\$f/g;s/T/\\$T\\$f/g")
else
foo=$(paste -d "" newsq* | sed 's/.\{3\}/& /g' | nl -v "$2" \
| sed "s/A/\\$A\\$f/g;s/C/\\$C\\$f/g;s/G/\\$G\\$f/g;s/T/\\$T\\$f/g")
fi
echo -e "$foo"
rm canon sq* newsq* working
exit